Molecules
A Molecules is an array of 3D molecules with three components.
def __init__(self, pos, rot=None, features=None): ...
pos(numpy.ndarray) … positions of the molecules in nanometers.rot(scipy.spatial.transform.Rotation) … rotation angles of the molecules.features(polars.DataFrame) … scalar features of the molecules.
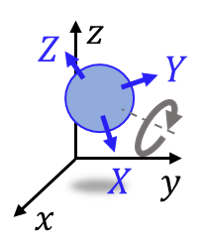
Fig. 1 Coordinate system of a molecule.
“Position” is a 3D coordinate of the molecules in nanometers. “Rotation” is a rotation operator that converts the molecule axes \(\vec{X}, \vec{Y}, \vec{Z}\) to the world coordinate axes \(\vec{x}, \vec{y}, \vec{z}\).
Hereafter, “world coordinate” means \(\vec{x}, \vec{y}, \vec{z}\) and “molecule coordinate” means \(\vec{X}, \vec{Y}, \vec{Z}\).
Note
Any arrays representing 3D coordinates are arranged in z, y, x order in acryo.
This is because the (x, y, z) element of a 3D array arr is accessed by
arr[z, y, x].
Note
acryo uses the right-handed coordinate system, which means that any axes
satisfy the rule \(\vec{x} \times \vec{y} = \vec{z}\). However, all the arrays
are in z, y, x order so that programatically, you have to calculate in the
left-handed manner.
For instance, if you have x, y vectors x and y, you have to run
z = -np.cross(x, y) to get the z vector. The rotation vector [np.pi, 0, 0]
corresponds to 90-degree anti-clockwise rotation around the z-axis.
Construction
The basic constructor of Molecules takes an array of positions and
another array that represents molecule rotations.
from scipy.spatial.transform import Rotation
from acryo import Molecules
mole = Molecules(
pos=[[0, 1, 2], [1, 2, 3]], # positions
rot=Rotation.from_euler('xyz', [[20, 0, 0], [0, 0, 0]]), # rotations
)
The mole object defined here has two molecules at positions \((z=0, y=1, x=2)\)
and \((z=1, y=2, x=3)\) with rotations given by the rot argument. The rotation
argument means that rot.apply([0, 0, 1]) matches the molecule axis \(\vec{X}\).
Many class methods defined in scipy.spatial.transform.Rotation are also
available as short-hand expressions. All of them take an array of positions and
other arguments required to construct a scipy.spatial.transform.Rotation.
Molecules.from_euler()… construction using Euler angles.Molecules.from_rotvec()… construction using rotation vector.Molecules.from_quat()… construction using quaternions.Molecules.from_matrix()… construction using rotation matrix.Molecules.from_random()… construction using random rotations.
mole = Molecules.from_euler(
pos=[[0, 0, 0], [1, 1, 1]],
angles=[[20, 0, 0], [0, 30, 0]],
degrees=True,
)
Physical Parameters
Physical parameters of Molecules can be obtained by following properties.
Molecules.pos… positions of molecules in a (N, 3) array.Molecules.rotator…scipy.spatial.transform.Rotationobject.
Array representation of the rotation can be obtained by following methods.
Molecules.euler_angle()Molecules.rotvec()Molecules.quaternion()Molecules.matrix()
Molecule Axes
The axes of the rotated molecules (\(\vec{X}, \vec{Y}, \vec{Z}\) in Fig. 1)
can be obtained as 3D vectors in the world coordinates using properties x, y and
z .
mole = Molecules.from_rotvec(
[[0, 0, 0]],
[[np.pi / 2, 0, 0]], # 90-degree rotation around z-axis
)
print(mole.x) # [0., -1., 0.]
print(mole.y) # [0., 0., 1.]
print(mole.z) # [1., 0., 0.]
Physical Transformation
Molecules supports several methods to transform molecules in the physical
coordinate system.
Lateral translation
If you want to translate molecules with their rotation fixed, following methods will be useful.
Molecules.translate()… translate molecules in the world coordinates.Molecules.translate_internal()… translate molecules in the molecule coordinates.
mole = Molecules([[0, 0, 0], [1, 1, 1]])
print(mole.pos) # [[0., 0., 0.], [1., 1., 1.]]
mole.translate([[1, 0, 0], [3, 3, -1]])
print(mole.pos) # [[1., 0., 0.], [4., 4., 0.]]
Self-centered rotation
If you want to rotate each molecules with their positions fixed, following methods will be useful.
Molecules.rotate_by()… rotate each molecule using ascipy.spatial.transform.Rotationobject.Molecules.rotate_by_euler_angle()… rotate each molecule using an array of Euler angles.Molecules.rotate_by_rotvec()… rotate each molecule using an array of rotation vectors.Molecules.rotate_by_quaternion()… rotate each molecule using an array of quaternions.Molecules.rotate_by_matrix()… rotate each molecule using an array of rotation matricesMolecules.rotate_by_rotvec_internal()… rotate each molecule using an array of rotation vectors. The components of the rotation vectors are described in the molecule coordinates of each molecules.
Molecule Features
“Features” means any scalar values associated with each molecule. Typical examples are:
The shift of each molecule from the original position after subtomogram alignment.
The cross-correlation coefficient between the subtomogram around each molecule and the reference image.
Cluster labels of each molecule after classification.
Molecules object has a property features that stores the features as a
polars.DataFrame object. You can set any DataFrame-like object to features.
# set features on construction
mole = Molecules(
pos=[[0, 0, 0], [1, 1, 1]],
features={'xcorr': [0.8, 0.9]},
)
# set features afterwhile
import polars as pl
mole.features = pl.DataFrame({'xcorr': [0.8, 0.9]})
Filter molecules
Molecule features can be used to filter molecules. The Molecules.filter() method
is a simple wrapper of polars.DataFrame.filter() to filter molecules by its features.
import polars as pl
mole = Molecules(
pos=[[0, 0, 0], [1, 1, 1], [2, 2, 2]],
features={'xcorr': [0.8, 0.9, 0.7]},
)
# filter molecules with xcorr > 0.85
mole_filt = mole.filter(pl.col('xcorr') > 0.85)
print(mole_filt.pos) # [[1., 1., 1.]]
Group molecules
Molecule features can be used to group molecules. The Molecules.groupby() method
is a simple wrapper of polars.DataFrame.groupby() to split a Molecules
object into sub-groups.
import polars as pl
mole = Molecules(
pos=[[0, 0, 0], [1, 1, 1], [2, 2, 2]],
features={"labels": ["A", "B", "A"]},
)
# group molecules by their labels
for name, mole_sub in mole.groupby("labels"):
print("label =", name)
print(mole_sub.pos)
# --- Out ---
# label = A
# [[0. 0. 0.]
# [2. 2. 2.]]
# label = B
# [[1. 1. 1.]]
Save Molecules
A Molecules object can be saved to a file using Molecules.to_csv() method.
This method merges the molecule positions, rotation and the features into a single table
data like below. In acryo, rotation vector is used to save the rotations because
it is the most compact form and is not as coordinate sensitive as Euler angle.
mole = Molecules.from_rotvec(
[[1, 2, 0], [3, 4, 1], [5, 6, 2]],
[[0.5, 0.1, 0.7], [0.6, 0.2, 0.4], [0.7, 0.3, 0.1]]
)
mole.to_csv("path/to/molecules.csv")
z |
y |
x |
zvec |
yvec |
xvec |
|---|---|---|---|---|---|
1.0 |
2.0 |
0.0 |
0.5 |
0.1 |
0.7 |
3.0 |
4.0 |
1.0 |
0.6 |
0.2 |
0.4 |
5.0 |
6.0 |
2.0 |
0.7 |
0.3 |
0.1 |