Subtomogram Loader
The main classes that actually perform subtomogram analysis are called “subtomogram loaders”.
A subtomogram loader is a pair of image(s) and a Molecules object, with
efficient mothods for loading, averaging or aligning subtomograms.
Currently, there are three subtomogram loaders in acryo.
SubtomogramLoader
A subtomogram loader that loads subtomograms from a single tomogram image.
BatchLoader
A subtomogram loader that loads subtomograms from multiple pairs of a tomogram image and a
Moleculesobject.MockLoader
A subtomogram loader that generates mock subtomograms.
These loaders have the same API. Here, I start with the SubtomogramLoader class to
show the basic usage of subtomogram loaders.
Creating a SubtomogramLoader
A SubtomogramLoader is a pair of a 3D tomogram image and a
Molecules object, with some additional parameters.
def __init__(self, image, molecules, order=3, scale=1.0, output_shape=Unset(), corner_safe=False): ...
image(numpy.ndarray or dask.Array) … the tomogram image.molecules(Molecules) … molecules in the tomogram.order(int) … order of the spline interpolation. 0=nearest, 1=linear, 3=cubic.scale(float) … scale (nm/pixel) of the tomogram image. This parameter must match the positions ofmolecules.output_shape(tuple) … shape of the output subtomograms, which will be used to determine the subtomogram shape during subtomogram averaging.corner_safe(bool) … if true, the subtomogram loader will ensure that the volume inside the given output shape will not be affected after rotation, otherwise the corners of the subtomograms will be dimmer.
SubtomogramLoader can be constructed from a image file using the imread() method
or the public imread() function.
from acryo import Molecules, imread
loader = imread("path/to/image.mrc", Molecules.from_csv("path/to/molecules.csv"))
Subtomogram Averaging
average() crops all the subtomograms around the molecules and
average them. This method always returns a 3D numpy.ndarray object.
from dask import array as da
from acryo import SubtomogramLoader, Molecules
image = da.random.random((100, 100, 100))
molecules = Molecules([[40, 40, 60], [60, 60, 40]])
# give output shape beforehand
loader = SubtomogramLoader(image, molecules, output_shape=(64, 64, 64))
avg = loader.average()
# or give output shape after construction
loader = SubtomogramLoader(image, molecules)
avg = loader.average(output_shape=(64, 64, 64))
Subtomogram Alignment
Templated alignment
align() crops all the subtomograms around the molecules and
align them to the given template image (reference image). This method will return
a new SubtomogramLoader object with the updated Molecules object.
You have to provide a template image, optionally a mask image, maximum shifts
in nanometers and an alignment model. The default alignment model is
ZNCCAlignment. For more details about the alignment models, see Alignment Model.
from dask import array as da
from acryo import SubtomogramLoader, Molecules
image = da.random.random((100, 100, 100))
template = np.random.random((20, 20, 20))
molecules = Molecules([[40, 40, 60], [60, 60, 40]])
loader = SubtomogramLoader(image, molecules)
out = loader.align(template, max_shifts=(5, 5, 5))
If you want to give parameters to the alignment model, you can use the with_params()
method of alignment model classes, or directly pass them to the **kwargs.
from acryo.alignment import ZNCCAlignment
loader = SubtomogramLoader(image, molecules)
# use with_params
out = loader.align(
template,
max_shifts=(5, 5, 5),
alignment_model=ZNCCAlignment.with_params(
rotations=[(6, 2), (6, 2), (6, 2)],
cutoff=0.5,
tilt=(-50, 50)
),
)
# directly pass them to the **kwargs
out = loader.align(
template,
max_shifts=(5, 5, 5),
alignment_model=ZNCCAlignment,
rotations=[(6, 2), (6, 2), (6, 2)],
cutoff=0.5,
tilt=(-50, 50),
)
Template-free alignment
If no a priori information is available for the template image, you’ll use the subtomogram
averaging result as the template image. During this task, each subtomogram will be loaded
twice so it is not efficient to call average() and align() separately.
align_no_template() creates a local cache of subtomograms so that alignment will be
faster.
loader = SubtomogramLoader(image, molecules)
out = loader.align_no_template(max_shifts=(5, 5, 5), output_shape=(20, 20, 20))
Multi-template alignment
If a tomogram is composed of heterogeneous molecules, you can use multiple templates to align the molecules and determine the best template for each molecule.
loader = SubtomogramLoader(image, molecules)
out = loader.align_multi_templates(
[template0, template1, template2],
max_shifts=(5, 5, 5),
label_name="template_id",
)
out.molecules.features["template_id"] # get the best template id for each molecule
Here, input templates must be given as a list of numpy.ndarray objects of the
same shape. label_name is the name used for the feature colummn of the best template.
Image preprocessing workflow
During subtomogram alignment, template images and mask images are usually provided from image files. They also need preprocessing such as rescaling and smoothing.
See Piping Images to the Loader for the details.
Filtering Loader
filter() is the method quite similar to that in Molecules or DataFrame.
It returns a new SubtomogramLoader object with the filtered molecules.
loader = SubtomogramLoader(image, molecules)
out = loader.filter(pl.col("score") > 0.5)
# all scores are greater than 0.5 after filtering
assert (out.molecules.features["score"] > 0.5).all()
This method is useful to filter out bad alignment,
loader.filter(pl.col("score") > 0.5)
choose molecules in certain regions,
loader.filter((10 < pl.col("x")) & (pl.col("x") < 20))
pick certain isotypes,
loader.filter(pl.col("cluster_id") == 1)
and so on.
Grouping Loader
Subtomogram loaders have a groupby() method. You can group molecules by a feature, create
corresponding subtomogram loaders and perform the same subtomogram analysis workflow efficiently.
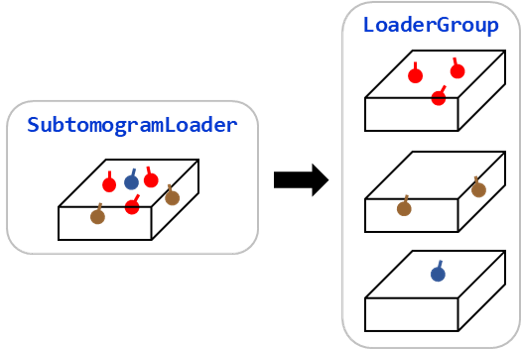
See Loader Group for the details.
Loading from Collection of Tomograms
Cryo-ET image analysis is usually performed on a collection of tomograms. Data management becomes very complicated in this case.
acryo provides a BatchLoader class for this purpose. BatchLoader
shares the same interface with SubtomogramLoader. It is constructed using the same parameters.
def __init__(self, order=3, scale=1.0, output_shape=Unset(), corner_safe=False): ...
BatchLoader can be constructed from a list of SubtomogramLoader objects.
from acryo import Molecules, imread, BatchLoader
collection = BatchLoader.from_loaders(
[
imread("path/to/image-0.mrc", Molecules.from_csv("path/to/molecules-0.csv")),
imread("path/to/image-1.mrc", Molecules.from_csv("path/to/molecules-1.csv")),
imread("path/to/image-2.mrc", Molecules.from_csv("path/to/molecules-2.csv")),
],
)
avg = collection.average(output_shape=(20, 20, 20))
out = collection.align(template, max_shifts=(5, 5, 5))
group = collection.groupby("cluster_id")
Mock Loader for Testing
MockLoader is for testing purpose only. The tomogram does not actually exist
but subtomograms are generated on the fly based on the template image. Subtomograms
are generated by following steps.
Affine transformation of the template image, based on the molecule position and rotation.
Calculate projections in different angles (Discrete Radon transformation).
Add noise to the projection.
Reconstruct the subtomogram (Weighted Back projection).
MockLoader is constructed using the following parameters.
def __init__(self, template, molecules, noise=0.0, degrees=None, central_axis=(0.0, 1.0, 0.0), ...): ...
template(numpy.ndarray or ImageProvider): template image that will be used to generate subtomograms.molecules(Molecules): pseudo molecules. The true center of the molecules is always at (0, 0, 0) and the true rotation is always the identity rotation. If you want to test shifting, say, [2, 3, 4], set the molecules position to [-2, -3, -4]. Same for rotation.noise(float): noise level. The noise is added to the projection of the template.degrees(float): tilt series rotation angles in degree.central_axis(tuple): central axis vector of the tilt series. The default is (0, 1, 0) which means the tilt series is rotated around the y-axis.